

Dra. Claudia Cannizzaro*
RESUMEN
Dentro de las patologías quirúrgicas del recién nacido, las oclusiones intestinales ocupan un lugar muy importante por su frecuencia. Comprenden un espectro amplio de patologías con un pronóstico variable.
Este artículo aborda la embriología, la clasificación, el diagnóstico prenatal y posnatal, el cuadro clínico y el tratamiento. En una segunda parte del artículo el Lic. David Apaza, se referirá a los cuidados de enfermería para los neonatos con oclusión intestinal.
Palabras clave: obstrucción intestinal, recién nacido, cirugía.
ABSTRACT
Among the surgical pathologies of the newborn, intestinal occlusions occupy a very important place be-cause of their frequency. They comprise a broad spectrum of pathologies with a variable prognosis.
This article addresses the embryology, classification, prenatal and postnatal diagnosis, clinical symptoms and treatment. In a second part of the article, BSN Da-vid Apaza will refer to nursing care for neonates with intestinal occlusion.
Keywords: intestinal obstruction, newborn, surgery.
Cómo citar:
Cannizzaro C. Oclusiones intestinales en el recién nacido. Parte1. Rev Enferm Neonatal. Abril 2019;29:8-21.
INTRODUCCIÓN:
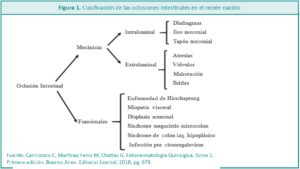
Dentro de las patologías quirúrgicas del recién nacido (RN), las oclusiones intestinales (OI) ocupan un lugar muy importante por su frecuencia. Representan un desafío para el equipo de salud, en referencia al diagnóstico precoz y la resolución clínico-quirúrgica, con el mínimo impacto posible para el desarrollo del niño.
Los avances en el diagnóstico prenatal, la minimización de las técnicas, el instrumental quirúrgico y la mejora en los cuidados intensivos neonatales, en especial por la calidad de la nutrición parenteral para neonatos, han hecho posible un aumento sostenido en la sobrevida y en la calidad de vida de niños cada vez más inmaduros y con malformaciones asociadas.1


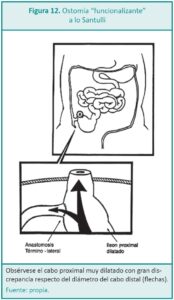
")


")




